Mission Bio's Multiple Myeloma (MM) Pipeline allows customers to process single-cell MM DNA and DNA + Protein sequencing data generated on the Tapestri Platform.
Table of Contents
Setting up Tapestri Pipeline Account
Single Cell Multiple Myeloma Sample Analysis Pipeline
MM DNA/DNA+P single sample analysis
MM DNA/DNA+P multiplexed sample analysis
Setting up Tapestri Pipeline Account
Refer to the Tapestri Pipeline User Guide to set up an account and access to Tapestri Pipeline.
Single Cell Multiple Myeloma Sample Analysis Pipeline
Mission Bio’s single cell Multiple Myeloma (scMM) analysis pipeline is a complete end-to-end solution for the analysis of Multiple Myeloma clinical samples. The pipeline enables assessing the presence of somatic clones based on mutations, whole genome and gene specific CNVs relative to a spike-in cell line (e.g. GM12878), VDJ clonotypes, and (optionally), cell-surface protein expression. Mutational co-occurrence and zygosity can be measured to understand clonal evolution. The pipeline provides a summary report and intermediate files with pertinent single-cell data. The pipeline is compatible with either a single sample or multiple samples multiplexed together that are distinguishable via their germline genotype information (which must also be provided). This end-to-end solution enables users to quickly assess the heterogeneity of thousands of cells in MM samples.
There are four MM analysis pipelines:
- MM DNA Pipeline
- MM DNA+Protein Pipeline
- MM Reprocess Pipeline
- Time course Pipeline
MM DNA Pipeline
MM DNA pipeline requires FASTQ files from a Tapestri DNA run (either from one sample, or from up to three multiplexed samples) and generates an MM report for each of the samples contained in the run.
MM DNA+Protein Pipeline
MM DNA+Protein pipeline requires FASTQ files from a Tapestri DNA+Protein run (either from one sample, or from up to three multiplexed samples) and generates an MM report for each of the samples contained in the run.
MM Reprocess Pipeline
MM Reprocess pipeline requires an h5 file from an existing MM DNA or MM DNA+Protein run and generates an MM report for each of the samples contained in the run. The Reprocess pipeline is used to run only the MM module which includes the DNA variant analysis, CNV analysis, VDJ clonotyping, and reporting. It should be used to resolve issues such as incorrect demultiplexing, whitelisting expected variants, or blacklisting unwanted or false positive variants.
Time course Pipeline
MM Time course analysis requires the h5 files from 2-5 existing MM DNA or MM DNA+Protein runs to generate a consolidated report summarizing the changes in the samples over time.
MM Inputs
MM pipeline has the following inputs:
FASTQ files
Input FASTQ files are one or more pairs of forward and reverse FASTQ files (R1/R2). These files should be compressed (.gz). DNA FASTQs are required for the MM DNA Pipeline, and DNA and Protein FASTQs are required for the MM DNA+Protein Pipeline.
Panel files
DNA panel files are required by the MM DNA Pipeline, and DNA and Protein panel files are required by the MM DNA+Protein Pipeline.
DNA Panel
The DNA panel consists of five files -
- *.bed
- *.amplicons
- systematic_variants.blacklist
- *.per-variant-background-error.csv
- *.amplicon.info.csv
The MM catalog DNA panel file can be found pre-uploaded in Tapestri Pipeline (Files → Panel Files). For more information about these panel files, refer to this article.
Protein Panel
The protein panel is supplied as a single csv file detailing the antibodies and their barcode sequences. The details of this panel file can be found here. The MM catalog Protein panel file can be found pre-uploaded in Tapestri Pipeline(Files → Panel Files).
Reference Genome
The Mission Bio-provided hg19 reference genome should be used for processing MM data. This catalog reference genome can be found pre-uploaded in Tapestri Pipeline (Files → Other Files).
CSV Files
MM Pipeline runs can include the following CSV files:
- Demultiplexing variants file (required for multiplexed DNA or DNA+Protein runs)
- Spike-in variants file (required for CNV detection)
- Whitelist/Blacklist variants file (optional)
- Spike-in CNV profile file (optional)
For more information about these input files, refer to this article.
Uploading CSV Files
Create the CSV file based on the details mentioned above, and then upload the file to Tapestri Pipeline. The CSV file must be uploaded before it can be used in a run. To upload a CSV file follow the instructions below:
- Click the Add Files button.
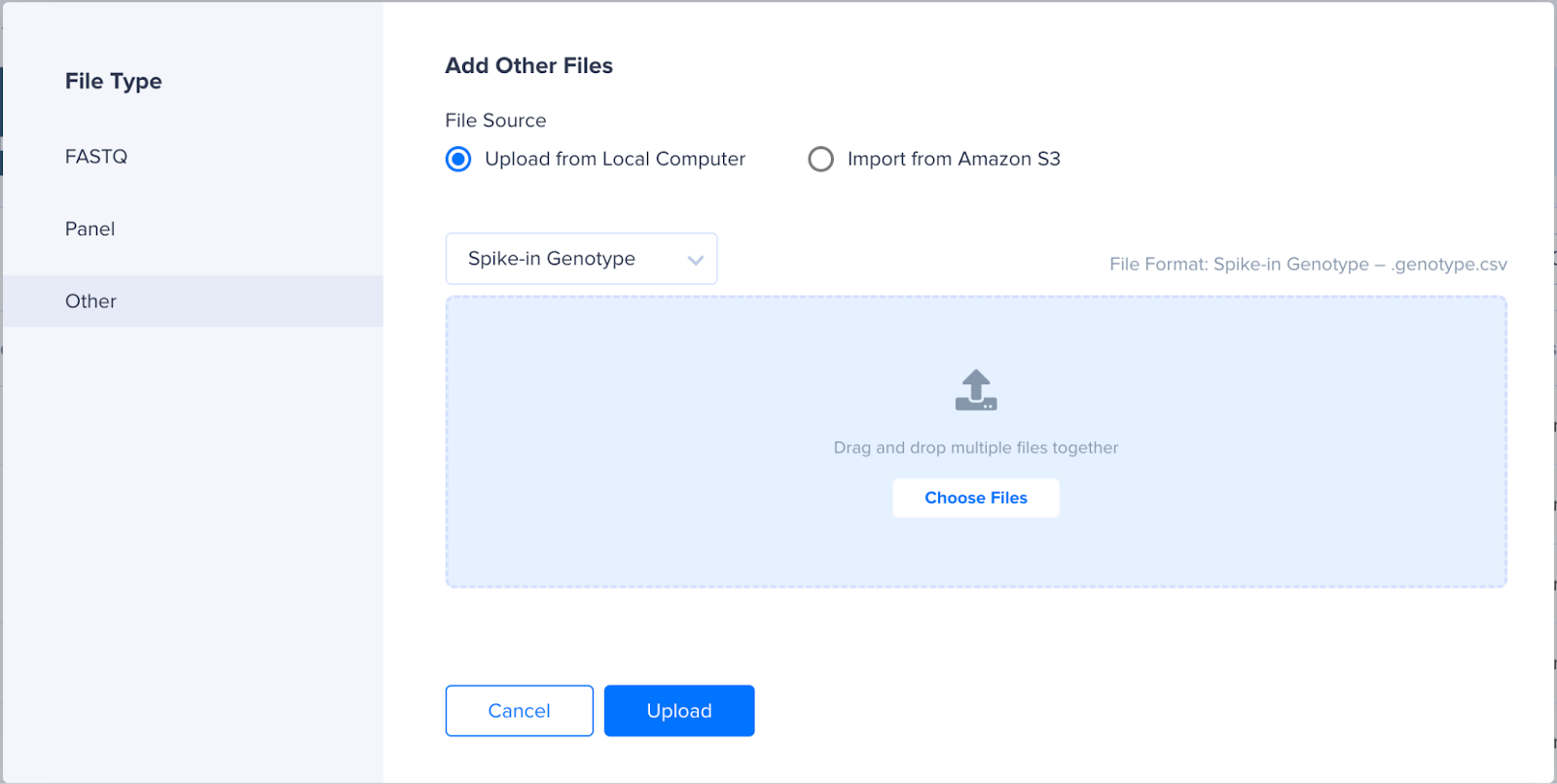
- Select the option Other from the Left panel.
- Choose either Upload from Local Computer or Import from Amazon S3 based on where the CSV files are saved.
- In the dropdowns, select the required type.
- Sample Variants File - To be used to upload the demultiplexing variant file. File extension is .csv.
- Somatic Variants File - To be used to upload the whitelisted/blacklisted variant file. File extension is .csv.
- Spike-in Genotype File - To be used to upload the spike-in variant file. File extension is .genotype.csv. If not provided, CNVs will not be called.
- Spike-in CNV File - To be used to upload the spike-in CNV file. File extension is .cnv.csv. If not provided, CNVs will be called with diploid assumption for the spike-in.
- Choose the files to add and click Upload.
- Once the upload completes, the files can be seen in the Other Files tab on the Files table.
To upload the FASTQ files, follow the same steps but select the File Type of FASTQ. Additional details on File source and configuring an AWS or Basespace account can be found here.
Starting MM Runs
The Tapestri Pipeline web application allows you to start four types of MM pipeline runs:
- MM DNA
- MM DNA+Protein
- MM Reprocess
- MM Time course
MM DNA
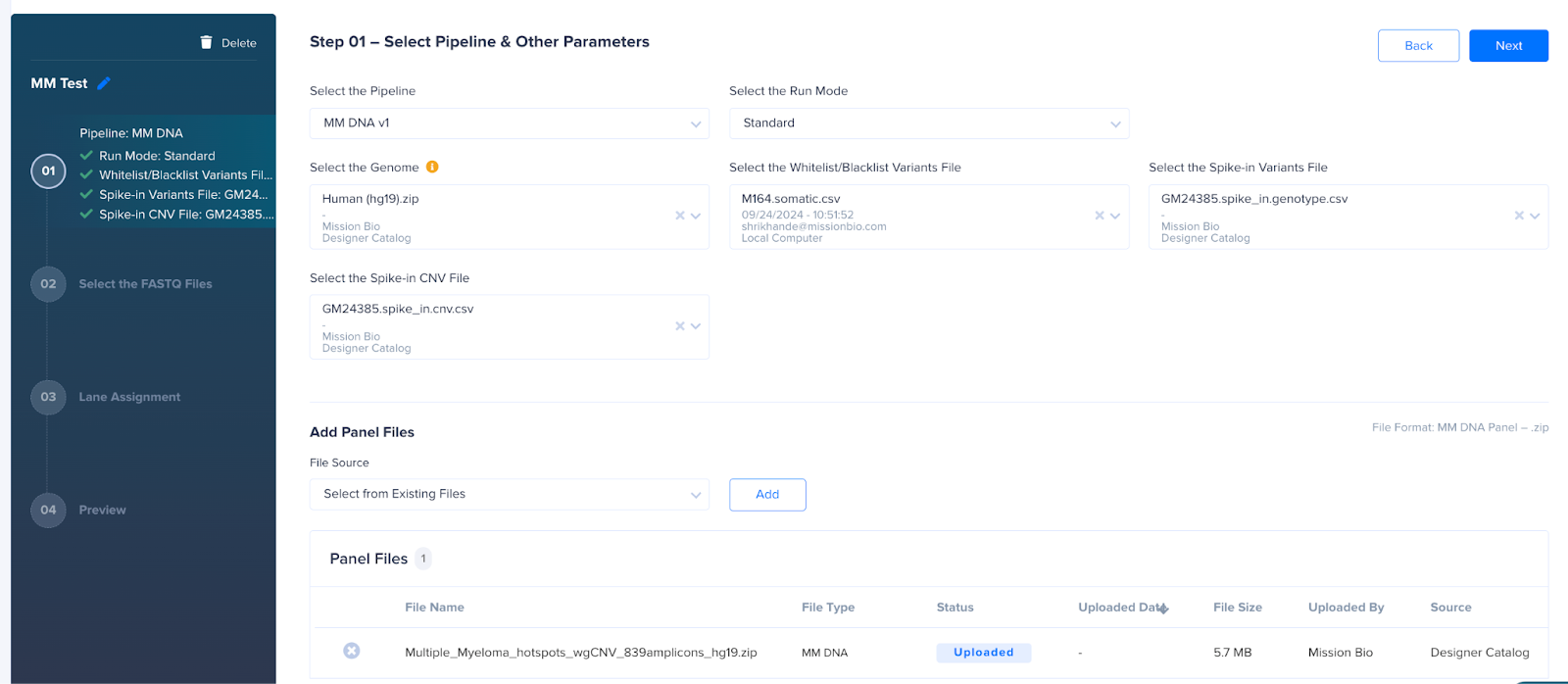
To process an MM DNA run, follow the steps given below:
- Click the Start Run button.
- Add the run name.
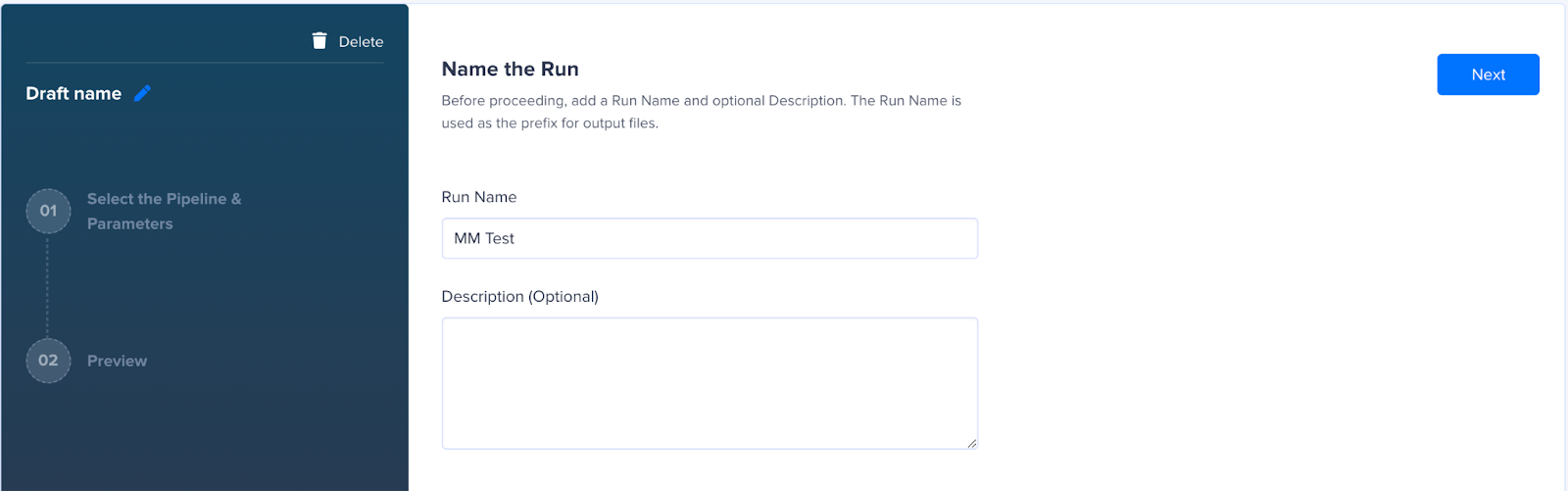
- Select the Pipeline MM DNA v2.
- Select the Human (hg19) genome.
- Select the Run Mode - Standard for single sample, Genotype Demultiplexing for multiplexed sample.
- Select the MM DNA Panel - Multiple_Myeloma_hotspots_339amplicons_hg19.zip if using the MM panelMultiple_Myeloma_hotspots_wgCNV_839amplicons_hg19.zip if using a combined MM+wgCNV panel.
- [Optional] Select the Whitelist/Blacklist Variants file for the run to define true and false positive variants to be included or excluded from analysis.
- [Optional] Select the Spike-in Variants file if the CNV calling is needed.
- [Optional] Select the Spike-in CNV file if the spiked-in cell line is not fully diploid.
- For Genotype Demultiplexing runs, additionally select the Demultiplexing Variants file.
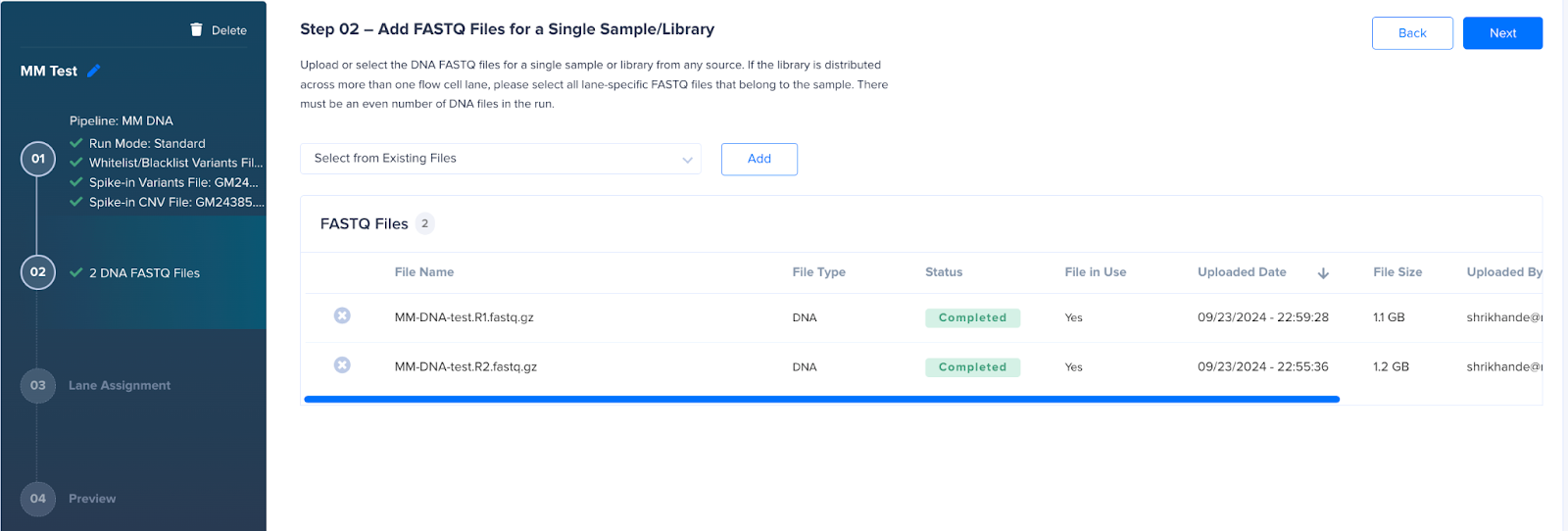
- Select the FASTQ files and assign them to correct lanes corresponding to your Tapestri experiment. See Lane assignment article for details.
- Preview the run inputs and submit the run.
- To view the results, click the name of the run in the Runs table.
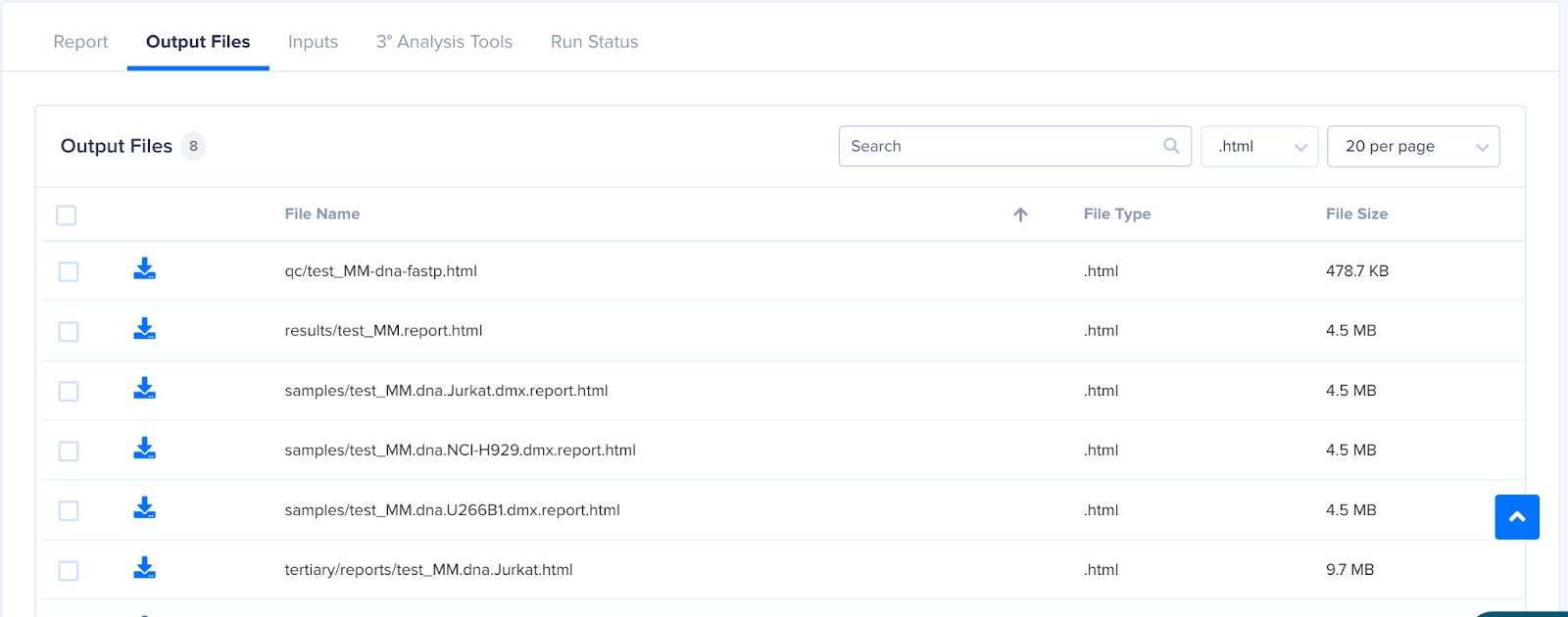
- The Run details page shows the run summary with Run Report, Output Files and Input Files. By default, the DNA pipeline report is seen on the Run Report tab.
- To view the MM reports, go to the Output Files tab and download the file tertiary/reports/{sample_name}.html.
MM DNA+Protein
To process an MM DNA+Protein run, follow the steps given below:
- Click the Start Run button.
- Add the run name.
- Select the Pipeline MM DNA+Protein v2
- Follow the same steps as for MM DNA run, with the following updates:
- While selecting parameters, select the catalog protein panel - TotalSeq™-D Multiple Myeloma Antibody Cocktail.csv
- In the next step select the Protein FASTQ files and assign them to the lanes correctly.
- To view the MM reports, go to the Output Files tab and download the file tertiary/reports/{sample_name}.html.
MM Reprocess
The MM Reprocess pipeline is used to run only the MM module, which includes the DNA variant analysis, CNV analysis, VDJ clonotyping, and reporting. It should be used to resolve issues such as incorrect demultiplexing, whitelisting expected variants, or blacklisting unwanted or false positive variants after the first run.
To start a MM Reprocess run, follow the steps below:
- Click the Start Run button.
- Add the run name.
- Select the Pipeline MM Reprocess v2.
- Select the Run Mode - Standard for single sample, Genotype Demultiplexing for multiplexed sample.
- Select the appropriate DNA panel.
NOTE: A protein panel file is not needed for this pipeline.
- [Optional] Select the Whitelist/Blacklist Variants file for the run to define true and false positive variants to be included or excluded from analysis.
- [Optional] Select the Spike-in Variants file if the CNV calling is needed.
- [Optional] Select the Spike-in CNV file if the spiked-in cell line is not fully diploid.
- For Genotype Demultiplexing runs, additionally select the Demultiplexing Variants file.
- Select the h5 file to be reprocessed.
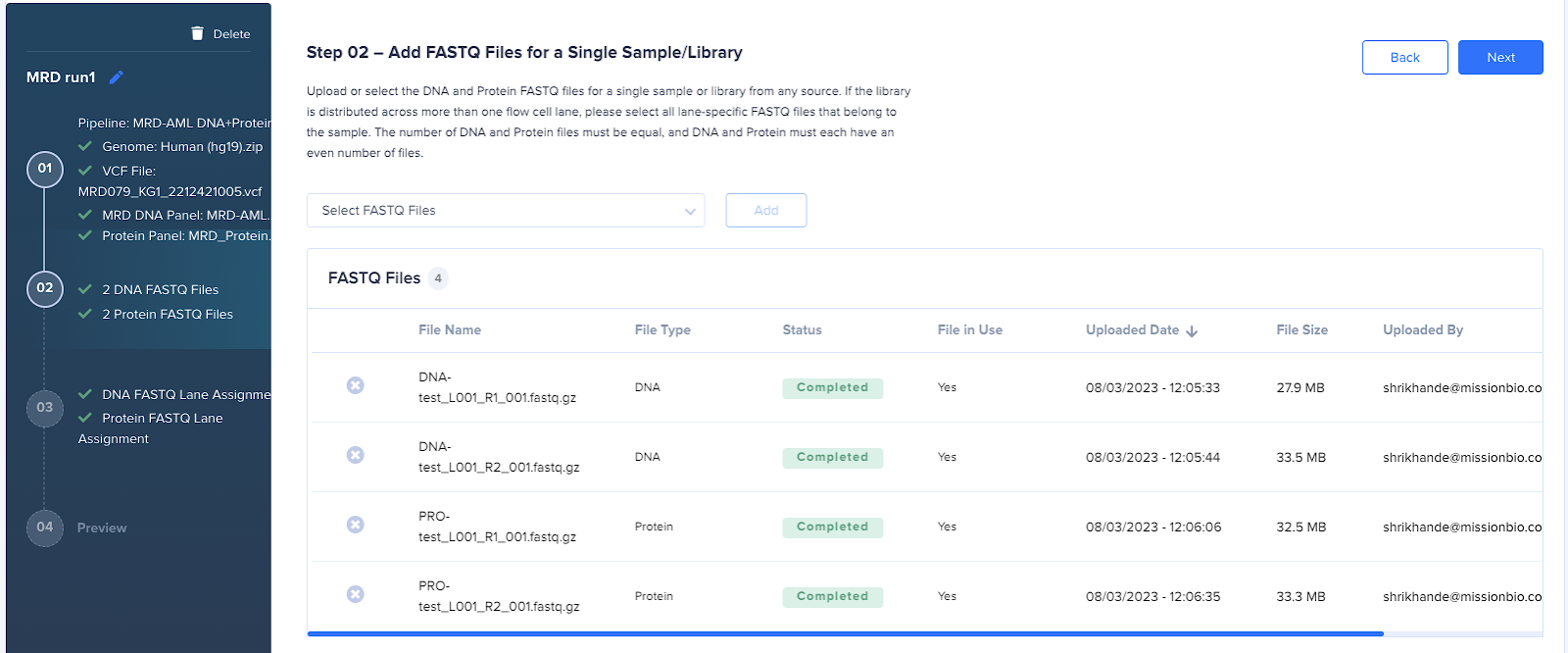
Note: For each run there are multiple h5s available; in order to select the correct file, first search the table using the output prefix. For example, if the run was named “MM test” then search for “MM_test” to limit the available h5 files. Once the h5 files are filtered, look for the one which is the output of the MM DNA or MM DNA+Protein pipeline. For MM DNA runs, the file name contains ‘results/’ and for DNA+Protein, the file name has no path or “/” in it. This is important as these h5 files contain the full assays (including the samples and the spike-in) and are unprocessed. Using other files may cause the run to fail.
- Preview the run inputs and submit the run.
- To view the results, click the name of the run in the Runs table.
- The Run details page shows the run summary with Run Report, Output Files and Input Files. By default, no report is seen on the Run Report tab as the secondary pipeline is not run in this process.
- To view the MM reports, go to the Output Files tab and download the file reports/{sample_name}.html.
MM Time course
This pipeline is used to combine patient samples across multiple time points. If you want to analyze a single patient over a period of time, then you can run the samples individually through the MM single sample pipeline and then use the h5 files from these runs to create a time course analysis report. To define the run, follow the steps below:
- Click the Start Run button.
- Add the run name.
- Select the Pipeline MM Time course.
- Select the appropriate DNA panel.
- Select the h5 files from previous MM runs listed in the table.
Note: For each run there are multiple h5s available; in order to select the correct file, first search the table using “samples/” in the name. This is important as these h5 files are processed and contain individual samples (and not multiplexed data). Using other files may cause the run to fail.
- Define the order or time point for the samples. There are 2 ways to define the order:
- Order the h5 files by the sequence in which the samples were collected. For example, the sample collected first can be assigned as 1, the next one as 2, the third one as 3, and so on.
- Specify the duration between the sample collection time points. For example, the first sample can be assigned as 1, a sample collected 20 days after that as 20, a sample collected 150 days later as 150, and so on.
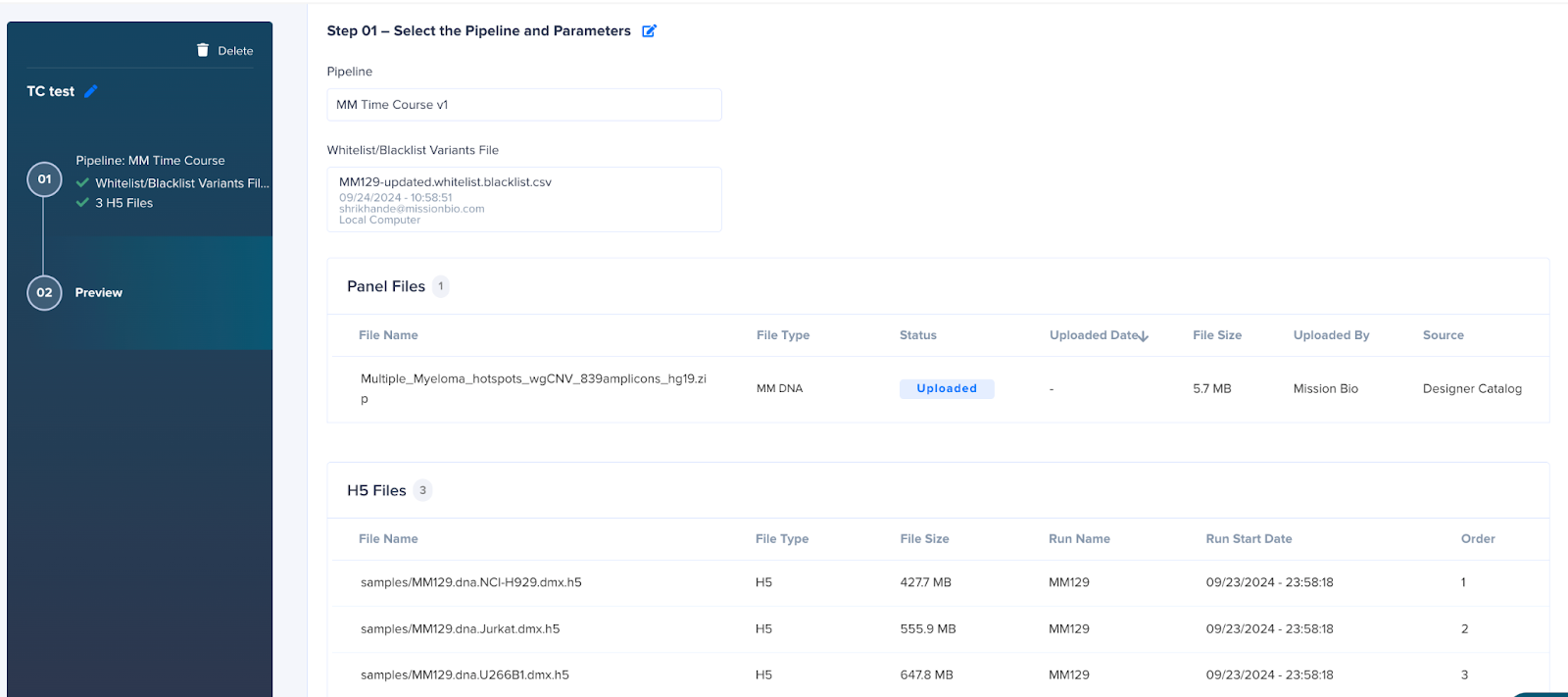
- Preview the run inputs and submit the run.
- To view the results, click the name of the run in the Runs table.
- The Run details page shows the run summary with Run Report, Output Files and Input Files. By default, the MM report is seen on the Run Report tab.
MM Output Files
MM pipeline outputs the following files:
MM DNA/DNA+P single sample analysis
MM pipeline executes secondary and tertiary analysis pipelines together; based on the modules run, different sets of files are generated:
-
Secondary analysis pipeline
- <prefix>.dna.h5 (DNA only) / dna+protein.h5 (DNA + Protein only)
- <prefix>.all.barcode.distribution.tsv.zip (DNA, DNA + Protein)
- <prefix>.cell.barcode.distribution.tsv.zip (DNA, DNA + Protein)
- tapestri_run_output.txt
- <prefix>.qc.json
- <prefix>-<analyte>-fastp.json and <prefix>-<analyte>-fastp.json
- <prefix>-<analyte>-fastp.html and <prefix>-<analyte>-fastp.html
- <prefix>.mapped.bam
- <prefix>.cells.bam
- <prefix>.cells.bam.csi
- <prefix>.report.html
- <prefix>.metrics.json
- <prefix>.cells.vcf.gz
- <prefix>.allele.drop.out.report.txt - Only Standard Run Mode
-
MM Specific Outputs
- samples/<prefix>.sample.report.html
- samples/<prefix>.sample.metrics.json
- samples/<prefix>.sample.h5
- samples/<prefix>.sample.cells.bam
- samples/<prefix>.sample.cells.bam.csi
- samples/<prefix>.<spikein_name>.spikein.report.html
- samples/<prefix>.<spikein_name>.spikein.metrics.json
- samples/<prefix>.<spikein_name>.spikein.h5
- samples/<prefix>.<spikein_name>.spikein.cells.bam
- samples/<prefix>.<spikein_name>.spikein.cells.bam.csi
- tertiary/reports/<prefix>.{sample}.html
- tertiary/h5/{sample}.h5
-
VDJ Specific Outputs
- vdj/<prefix>_<bcr_type>-<gene>_summary.tsv
- vdj/<prefix>_<bcr_type>-<gene>_summary_filtered.tsv
- vdj/<prefix>_report.tsv
- vdj/<prefix>_report_filtered.tsv
- vdj/<prefix>_metrics.json
- vdj/logs/progress.log
MM DNA/DNA+P multiplexed sample analysis
MM pipeline executes secondary and tertiary analysis pipelines together; based on the modules run, different sets of files are generated:
-
Secondary analysis pipeline
- <prefix>.dna.h5 (DNA only) / dna+protein.h5 (DNA + Protein only)
- <prefix>.all.barcode.distribution.tsv.zip (DNA, DNA + Protein)
- <prefix>.cell.barcode.distribution.tsv.zip (DNA, DNA + Protein)
- tapestri_run_output.txt
- <prefix>.qc.json<prefix>-<analyte>-fastp.json and <prefix>-<analyte>-fastp.json
- <prefix>-<analyte>-fastp.html and <prefix>-<analyte>-fastp.html
- <prefix>.mapped.bam
- <prefix>.cells.bam
- <prefix>.cells.bam.csi
- <prefix>.report.html
- <prefix>.metrics.json
- <prefix>.cells.vcf.gz<prefix>.allele.drop.out.report.txt - Only Standard Run Mode
-
MM Specific Outputs
- samples/<prefix>.<spikein_name>.spikein.report.html
- samples/<prefix>.<spikein_name>.spikein.metrics.json
- samples/<prefix>.<spikein_name>.spikein.h5
- samples/<prefix>.<spikein_name>.spikein.cells.bam
- samples/<prefix>.<spikein_name>.spikein.cells.bam.csi
-
Multiple copies of following files, one set for each multiplexed sample and a single set of files for spike-in cell line:
- tertiary/reports/<prefix>.{sample}.html
- tertiary/h5/{sample}.h5
- samples/<prefix>.{sample}.dmx.report.html
- samples/<prefix>.{sample}.dmx.metrics.json
- samples/<prefix>.{sample}.dmx.h5
- samples/<prefix>.{sample}.dmx.cells.bam
- samples/<prefix>.{sample}.dmx.cells.bam.csi
-
VDJ Outputs
- vdj/<prefix>_<bcr_type>-<gene>_summary.tsv
- vdj/<prefix>_<bcr_type>-<gene>_summary_filtered.tsv
- vdj/<prefix>_report.tsv
- vdj/<prefix>_report_filtered.tsv
- vdj/<prefix>_metrics.json
- vdj/logs/progress.log
MM Time Course analysis
MM time course analysis consolidates 2-5 sample H5s and creates the following output files:
- <prefix>.html
- <prefix>.h5
For more information about Multiple Myeloma output files, refer to this article.
To download any output file, click the download icon to the left of the File Name.
Note: if the file does not download, see if you have an ad popup blocker running. If so, disable it, and download the file again.
MM Report Overview
To download the MM Run Report, go to the Output Files tab and download the tertiary/reports/{sample_name}.html file. Plots and tables in the report are interactive.
Summary
The Summary page displays the following information:
- Total cells
- Mutant cells detected
- Mutant clones detected
- Clonal summary plot: Visual representation of Phylogeny, Clonal Fraction, VDJ Clonotypes, Prognostic Structural Variants, Structural Variant Count, Focal CNV, Point Mutations and Protein Cluster Fraction.
- Clones table: Table with clone name, number of cells, mutations, protein differential expression, large CNVs, focal CNVs and VDJ clonotypes.
- CNV correlated variants table: A table with the sample name (for time course), variant ID, gene, protein change, coding impact, cells mutated % and various other metrics.
Advanced
The Advanced page displays the following information:
- Phylogenetic tree: A visualization showing the order in which the mutations were acquired and how they co-occur.
- DNA & protein profile: A heatmap showing DNA Cluster Signature subsorted by Protein.
- CNV profile: A plot showing the genome-wide CNV events.
- Protein cluster profile: A plot showing the normalized expression level for each cluster for all antibodies which have expression above 0.5 for at least one cluster.
- Protein UMAP: A UMAP plot showing the protein expression colored by either Protein, sample, clone or genotype.
- Protein expression correlation: A plot showing the correlation in expression for two proteins.
- Protein expression change over time: A time course analysis-only plot showing the change in protein expression over a period of time.
- VDJ clonotypes: A bar chart that shows unique VDJ recombination events observed in the clones
- VDJ clones table: A detailed information for all unique VDJ recombination events observed in the sample.
-
Sample
- Run ID
- Sample ID
- DNA panel name
- DNA panel size
- Reference genome
- Secondary analysis pipeline version
- Tertiary analysis pipeline version
- Date analyzed
QC
- Raw CNV counts with VAFs: It shows the raw CNV counts with average variant allele frequency (VAF) in each clone for germline variants that are located in the corresponding amplicons.
- Heatmap of somatic variants (raw genotypes): A heatmap showing raw genotypes for the somatic variants per cell.
- Heatmap of protein expression: A heatmap showing the normalized protein expression per cell.
- Heatmap of VDJ read counts: A heatmap of the read counts for each VDJ gene segment in each cell.
- Candidate variants Table: If there are amplicons which cover somatic variants, then this table is shown. It contains detailed information for all variants that pass a lenient filtering criteria. For each variant, the table shows the number of cells mutated, the percentage of cells mutated, reason for filtering the variant, whether the variant is passed as a whitelist or as a germline variant, and multiple Varsome annotations. The filtering happens sequentially and the annotations are only shown for variants which pass all the preceding filtering criteria. If a variant is a known somatic variant (whitelisted), it might still contain a value in the "Reason" column. It shows the reason the variant would have been filtered if it were not whitelisted. This table can be used to identify why a particular variant was not called in the report and is especially useful in cases when the variant was missed as it was defined as a germline variant for demultiplexing purposes.
Definitions
The Definitions page contains a glossary of key words used in the report and a description of every table and plot contained in the report.