Basics
Without an account, anyone can navigate to https://designer.missionbio.com/catalog-panels to view catalog panels that come in ready-to-ship or ready-to-customize formats.
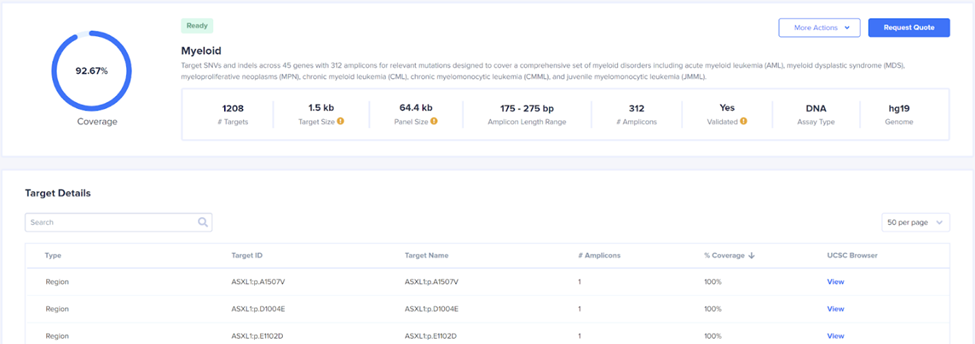
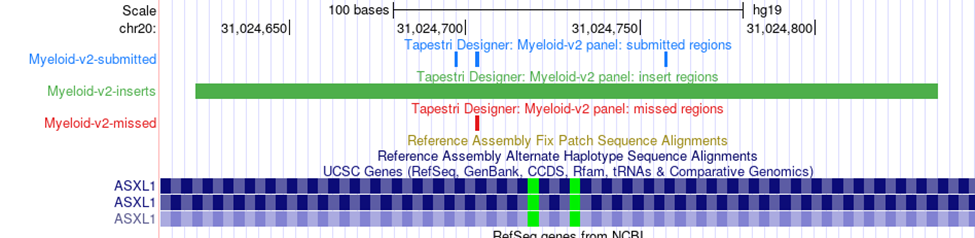
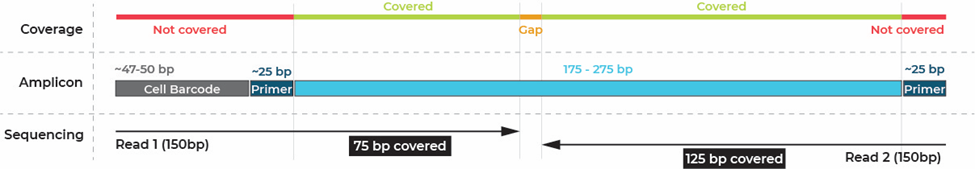
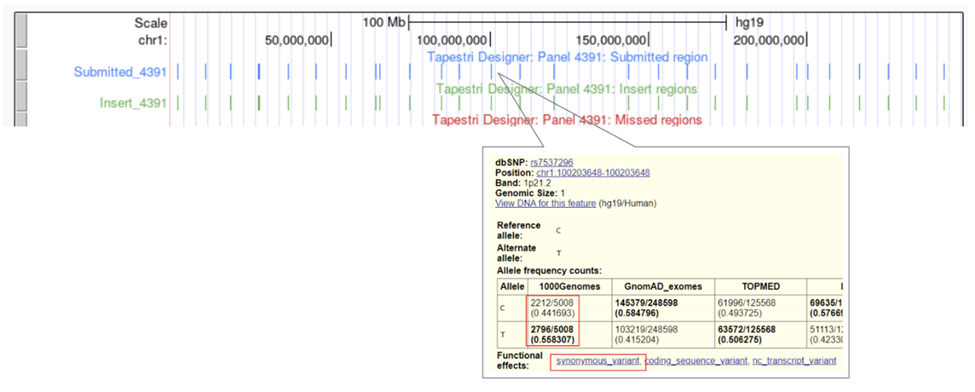
Viewing a panel shows basic information such as the number of amplicons, the percent coverage of the supplied targets, and a “More Actions” drop down menu where a data sheet or the panel .zip file can be downloaded. Two useful files within the zip are the coverage summary, which lists the submitted targets and their coverage, as well as the design summary, which shows the exact start and end points of each amplicon, insert, and primer in the panel. The following link reviews in detail the various files in the designer zip: Designer zip file. The blue View link after each target will take you to the UCSC browser with 3 tracks pre-loaded. A blue track indicating the location of the submitted targets, a green track showing the position of the insert (primers not included), and a red track showing the missed targets. Note that because of the cell barcode and primer at the beginning of the amplicon, some targets covered by the insert may still be missed using 2x150 bp sequencing. See the amplicons structure and example from the Myeloid panel below.
Creating Custom Panels
The specific steps in creating a custom panel are detailed at the following link: Creating a custom panel. This guide is designed to offer best practices and considerations when creating a panel.
Creating a target list
When using the built in hg19 genome, there are 4 target types that can be submitted, details here: Target types.
It is recommended to use a spreadsheet (.csv) to build a target list using the above format. This allows for easy manipulation of the targets should changes need to be made. Once all the targets have been gathered, they can be pasted into the submission window.
The Gene target type submits the CDS of all transcripts of a gene and is an easy way to quickly maximize coverage across an entire gene. If there are specific hotspots within a gene that need additional priority, they can be added in addition to the gene, such as in the table above for TP53.
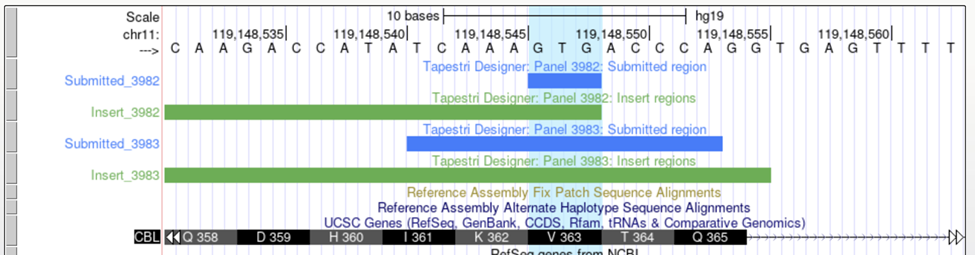
In practice, it can be easiest to use only the Gene and Region target types. Using the region target type has the benefit of being unambiguous, as opposed to dbSNP, which may inadvertently be from an outdated database version, or HGVS, which may target the wrong or multiple transcripts. Additionally, there is some evidence that the first 1-5 base pairs after the primer may have slightly reduced quality in amplicon based sequencing. Using region allows the addition of +/- 5 bp of padding to help prevent targets from residing at the very beginning or end of an insert. For example, submitting chr11:119148541-119148553 (tracks 3982) instead of chr11:119148546-119148548 (tracks 3983) to target CBL:p.V363.
When using custom reference genomes, hg38 (GRCh38) or mm10 only the Region target type is allowed
Targeting CNV events
Copy number can be inferred from the read depth across amplicons. Because all amplicons suffer from random allelic dropout to a certain extent, it is critical to have multiple amplicons in a given region to have confidence in identifying CNV. We recommend 5-6 amplicons across a gene or chromosome band, and 10-12+ amplicons across an entire chromosome arm. When possible, target high Minor Allele Frequency SNPs. If a sample happens to be Het, changes in the allele frequency can help identify loss of heterozygosity or allele gain. Amplicons already generated to target SNPs function for CNV as well.
Copying Panels
It is possible to copy a completed custom or catalog panel. This is useful when only a few targets need to be added or deleted as there is no function to batch add targets.

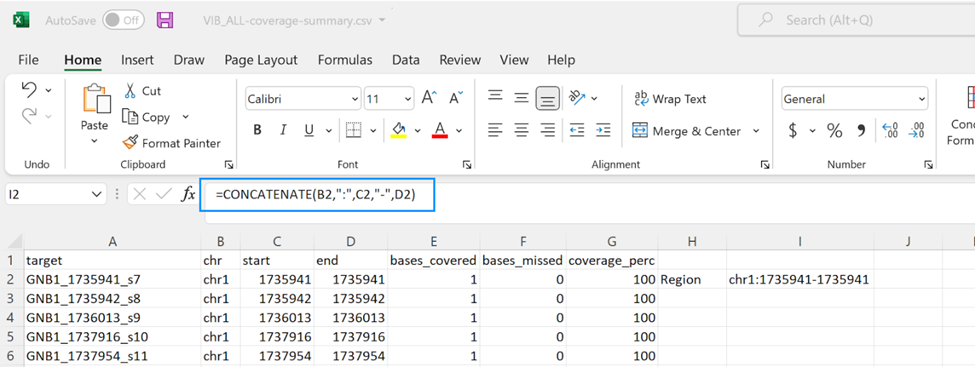
To make extensive changes to a panel, the coverage summary file can be used to quickly make a new target list (Try using =CONCATENATE(B2,":",C2,"-",D2) in Excel to get the correct format for submission of Region type targets).
Optimizing panel content
It can be useful to initially submit an entire target list to get a general idea on number of amplicons, overall coverage, and coverage of ‘must have’ targets. Generally, missed targets will belong to two broad categories. The first category is amplicon or target proximity related, and prioritization can often rescue ‘must have’ targets after removing low priority targets. The second category is related to the sequence of the target, and presents a challenge as any potential rescue is only possible by relaxing design criteria (e.g., GC percentage), which may impact amplicon performance.
When working to rescue missed targets, it can speed up the process to only work with the region close to the missed targets (e.g. a single gene or exon) so that the panel will be generated quickly and multiple iterations can be done in a small amount of time. Once the smaller region is optimized, the entire set of targets can again be submitted to generate the complete panel. Occasionally, distant primer sets can have negative interactions resulting in loss of coverage after optimizing a small region, but it is rare.
To rescue amplicon or target proximity related misses requires prioritization of the targets. One approach is to submit ‘must have’ targets on their own to make sure they can be covered when there are the least amount of conflicts, and then gradually add back in lower priority targets. Often viewing the targets in the UCSC browser can help identify how to rescue coverage. In the example below, removal of a target at the beginning of the left amplicon helps rescue a target at the end of the amplicon.
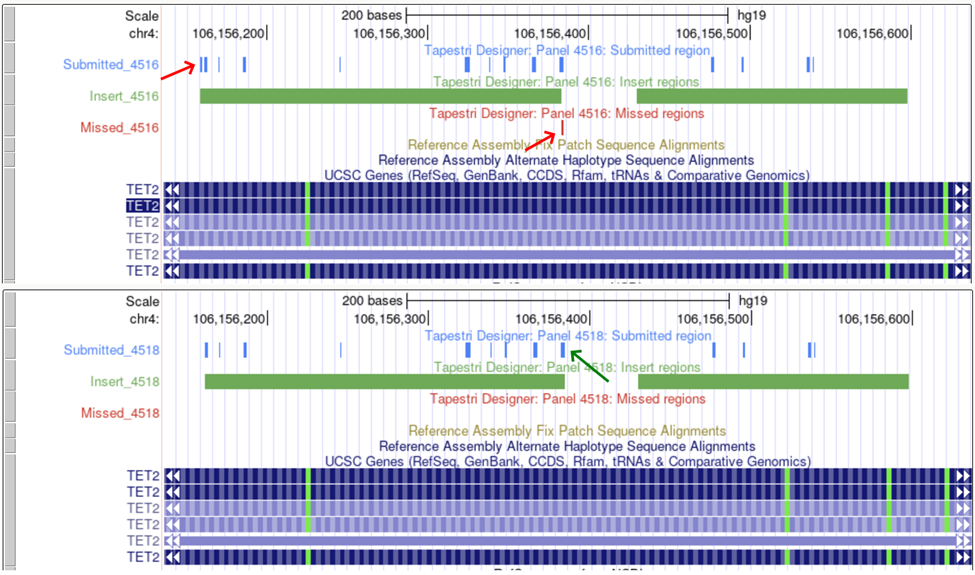
Some target regions cannot be easily recovered. UCSC can show several useful tracks that can help identify why some targets are missed.
High GC content
Tapestri Designer will design amplicons in regions with 27-70% GC content. Primers are restricted to 27-62% GC content. Regions outside this GC content are more difficult to amplify by PCR, and the corresponding amplicons are at higher risk to underperform (e.g., low number of reads). Amplicons generated with GC content 62-70% are shown with a flag in the final design to warn of possible poor performance.
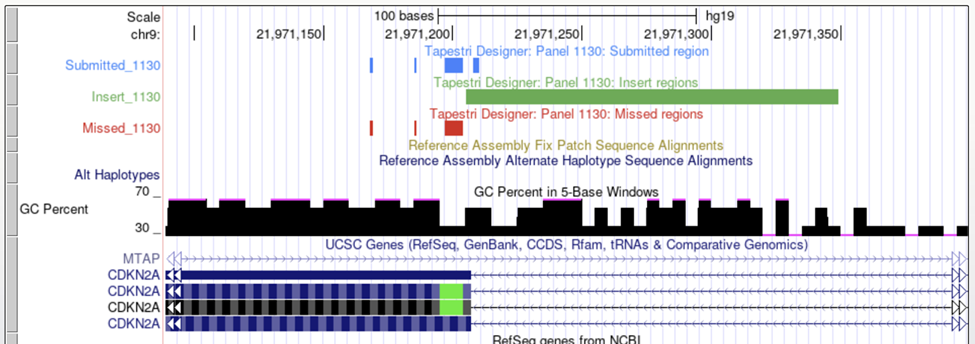
UCSC Visualization
Enabling the ‘GC Percent’ track can assist in identifying areas of high GC content. DNA sequences can easily be retrieved from UCSC by selecting ‘DNA’ from the ‘View’ menu and used to calculate exact GC percentages for a specific region.
How to address
GC content thresholds help maintain panel uniformity by ensuring amplicons will perform evenly during PCR. White Glove design can attempt to generate amplicons with up to 74% total GC content, but these amplicons may perform poorly and negatively impact downstream analysis. Targets in areas with GC content >74% cannot be rescued.
Masked Regions
Tapestri Designer cannot generate amplicons in masked regions of the genome. This is any area where the sequence is listed as an N (e.g., centromere regions, telomere regions). Having even a single target with a masked region may cause the entire panel to fail to generate, rather than show as missed coverage.
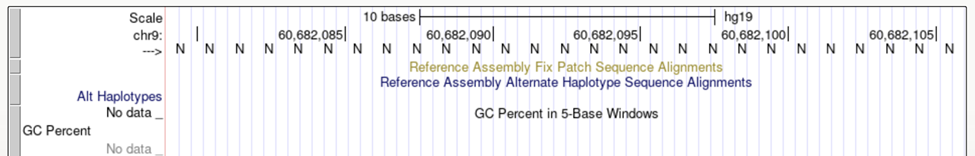
How to address
Masked regions may be rescued using a custom reference genome to supply missing sequence information.
Repetitive Regions
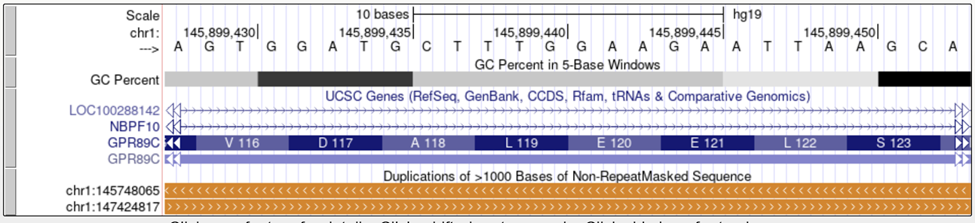
Tapestri Designer may not generate amplicons in highly repetitive regions. In the example below, there is a CAG repeat to the right of the target. The designer cannot generate a unique amplicon for this region.

UCSC Visualization
Enable the ‘RepeatMasker’ track to help identify repeat regions.
How to address
If a long repetitive region needs to be covered, White Glove design can assist in generating a panel using PE 250 chemistry to increase read/amplicon length.
Homologous Regions
Tapestri Designer may not generate amplicons in repeat elements with high homology. LINEs, SINEs, homologous genes, etc may contribute to this.
UCSC Visualization
Various tracks in the ‘Repeats’ menu can aid in identifying different types of repetitive regions.
How to address
Repetitive regions can be hard masked and uploaded as a custom reference genome. This approach is perhaps most useful for targeting genes duplicated in chrX/Y that cause Tapestri Designer to fail while typically only being present in 2 copies in cells. If designing an experiment with targets repeated in the genome, careful consideration should be taken in terms of panel performance and uniformity, sequencing, and downstream analysis.
Quick Tips
- It is recommended to add +/- 5 bp of padding to each SNV target to prevent them from being at the exact start or end of an exon.
- For CNV coverage it is recommended to have 5-6 amplicons on a gene or chromosome band level and 10-12+ on a chromosome arm level.
- Reach out to support@missionbio.com if including mitochondrial targets. The increased relative abundance of mtDNA influences the panel and experimental design.
-
The R2 read is typically on the right end of the amplicon and offers longer coverage due to the barcode sequence consuming the first 50 bp of R1. To optimize placement of the amplicon, increase the submitted target size. This can be useful to capture an indel or CRISPR target site in a single R2 read rather than have it split over a paired R1/R2.

Resources for Panel Content
There are many online databases of SNV and CNV alterations from various diseases
- COSMIC
- dbSNP
- The Genomic Data Commons - Access to The Cancer Genome Atlas data
- The International Cancer Genome Consortium